Cellular processes are intricately controlled through gene regulation, which is significantly influenced by intrinsic noise due to the small number of molecules involved. The Gillespie algorithm, a widely used stochastic simulation method, is pervasively employed to model these systems. However, this algorithm typically assumes that DNA is homogeneously distributed throughout the nucleus, which is not realistic. In this study, we evaluated whether stochastic simulations based on the assumption of spatial homogeneity can accurately capture the dynamics of gene regulation. Our findings indicate that when transcription factors diffuse slowly, these simulations fail to accurately capture gene expression, highlighting the necessity to account for spatial heterogeneity. However, incorporating spatial heterogeneity considerably increases computational time. To address this, we explored various stochastic quasi-steady-state approximations (QSSAs) that simplify the model and reduce simulation time. While both the stochastic total quasi-steady state approximation (stQSSA) and the stochastic low-state quasi-steady-state approximation (slQSSA) reduced simulation time, only the slQSSA provided an accurate model reduction. Our study underscores the importance of utilizing appropriate methods for efficient and accurate stochastic simulations of gene regulatory dynamics, especially when incorporating spatial heterogeneity.
Molecular glues are small molecules that offer a powerful strategy to target previously “undruggable” proteins of interest (POI) by enhancing their interactions with other proteins (effector). Depending on the nature of the effectors, molecular glues can induce stabilizing sequestration or degradation of POI. However, their rational design has been hindered by a poor understanding of how kinetic parameters impact their performance. To address this, we developed a unified mathematical framework that accurately analyzes the dynamics of both glue degraders and stabilizers. Our model reveals that the strong binding affinity of the ternary complex is the key determinant of performance across both modalities, provided that the initial component concentrations and degradation rate constants are fixed. Furthermore, we demonstrate that degrader performance is ultimately limited by its catalytic efficiency and the target protein’s natural half-life. We also identify distinct roles for effector abundance, showing that the relative concentrations of the effector and POI are critical for stabilizers but less so for degraders. This quantitative framework provides mechanistic principles for the rationale design and optimization of molecular glues.Competing Interest StatementS.M.C., G.G., J.T., M.C., R.R., J.W., and M.J. are present or former employees and may be shareholders of AstraZeneca. The remaining authors declare no competing interests.
All proteins are translated in the cytoplasm, yet many, including transcription factors, play vital roles in the nucleus. While previous research has concentrated on molecular motors for the transport of these proteins to the nucleus, recent observations reveal perinuclear accumulation even in the absence of an energy source, hinting at alternative mechanisms. Here, we propose that structural properties of the cellular environment, specifically the endoplasmic reticulum (ER), can promote molecular transport to the perinucleus without requiring additional energy expenditure. Specifically, physical interaction between proteins and the ER impedes their diffusion and leads to their accumulation near the nucleus. This result explains why larger proteins, more frequently interacting with the ER membrane, tend to accumulate at the perinucleus. Interestingly, such diffusion in a heterogeneous environment follows Chapman’s law rather than the popular Fick’s law. Our findings suggest a novel protein transport mechanism arising solely from characteristics of the intracellular environment.
High dimensionality and noise have limited the new biological insights that can be discovered in scRNA-seq data. While dimensionality reduction tools have been developed to extract biological signals from the data, they often require manual determination of signal dimension, introducing user bias. Furthermore, a common data preprocessing method, log normalization, can unintentionally distort signals in the data. Here, we develop scLENS, a dimensionality reduction tool that circumvents the long-standing issues of signal distortion and manual input. Specifically, we identify the primary cause of signal distortion during log normalization and effectively address it by uniformizing cell vector lengths with L2 normalization. Furthermore, we utilize random matrix theory-based noise filtering and a signal robustness test to enable data-driven determination of the threshold for signal dimensions. Our method outperforms 11 widely used dimensionality reduction tools and performs particularly well for challenging scRNA-seq datasets with high sparsity and variability. To facilitate the use of scLENS, we provide a user-friendly package that automates accurate signal detection of scRNA-seq data without manual time-consuming tuning.
The Michaelis–Menten (MM) rate law has been a fundamental tool in describing enzyme-catalyzed reactions for over a century. When substrates and enzymes are homogeneously distributed, the validity of the MM rate law can be easily assessed based on relative concentrations: the substrate is in large excess over the enzyme-substrate complex. However, the applicability of this conventional criterion remains unclear when species exhibit spatial heterogeneity, a prevailing scenario in biological systems. Here, we explore the MM rate law’s applicability under spatial heterogeneity by using partial differential equations. In this study, molecules diffuse very slowly, allowing them to locally reach quasi-steady states at each time point. We find that the conventional criterion for the validity of the MM rate law cannot be readily extended to heterogeneous environments solely through spatial averages of molecular concentrations. That is, even when the conventional criterion for the spatial averages is satisfied, the MM rate law fails to capture the enzyme catalytic rate under spatial heterogeneity. In contrast, a slightly modified form of the MM rate law, based on the total quasi-steady state approximation (tQSSA), is accurate. Specifically, the tQSSA-based modified form, but not the original MM rate law, accurately predicts the drug clearance via cytochrome P450 enzymes and the ultrasensitive phosphorylation in heterogeneous environments. Our findings shed light on how to simplify spatiotemporal models for enzyme-catalyzed reactions in the right context, ensuring accurate conclusions and avoiding misinterpretations in in silico simulations.
The circadian (∼24h) clock is based on a negative-feedback loop centered around the PERIOD protein (PER), translated in the cytoplasm and then enters the nucleus to repress its own transcription at the right time of day. Such precise nucleus entry is mysterious because thousands of PER molecules transit through crowded cytoplasm and arrive at the perinucleus across several hours. To understand this, we developed a mathematical model describing the complex spatiotemporal dynamics of PER as a single random time delay. We find that the spatially coordinated bistable phosphoswitch of PER, which triggers the phosphorylation of accumulated PER at the perinucleus, leads to the synchronous and precise nuclear entry of PER. This leads to robust circadian rhythms even when PER arrival times are heterogeneous and perturbed due to changes in cell crowdedness, cell size, and transcriptional activator levels. This shows how the circadian clock compensates for spatiotemporal noise.
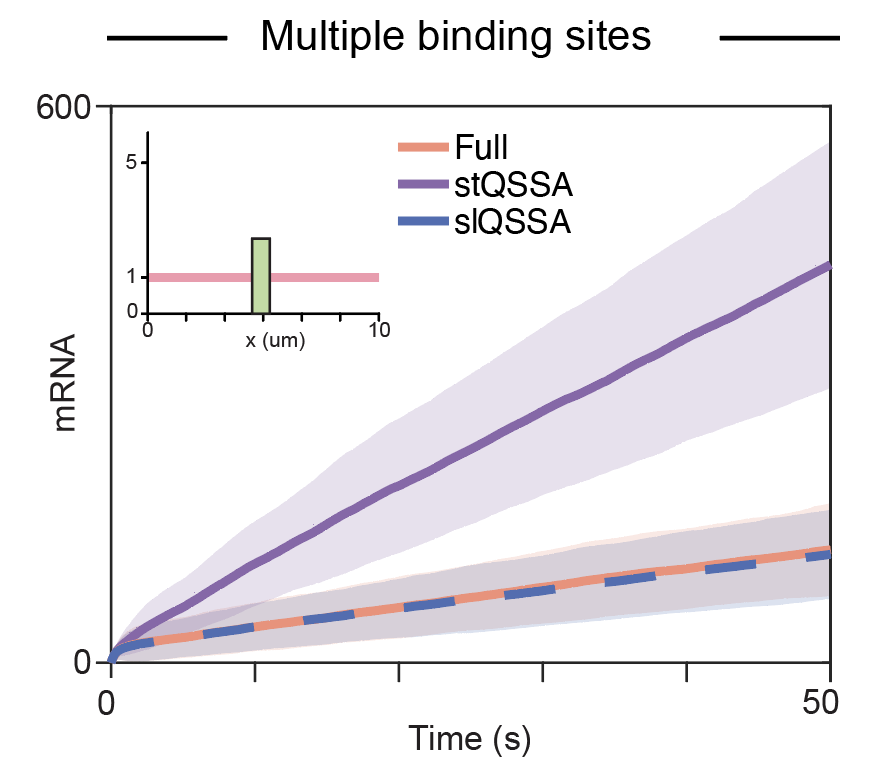 From Homogeneity to Heterogeneity: Refining Stochastic Simulations of Gene RegulationComputational and Structural Biotechnology Journal, 2025
From Homogeneity to Heterogeneity: Refining Stochastic Simulations of Gene RegulationComputational and Structural Biotechnology Journal, 2025
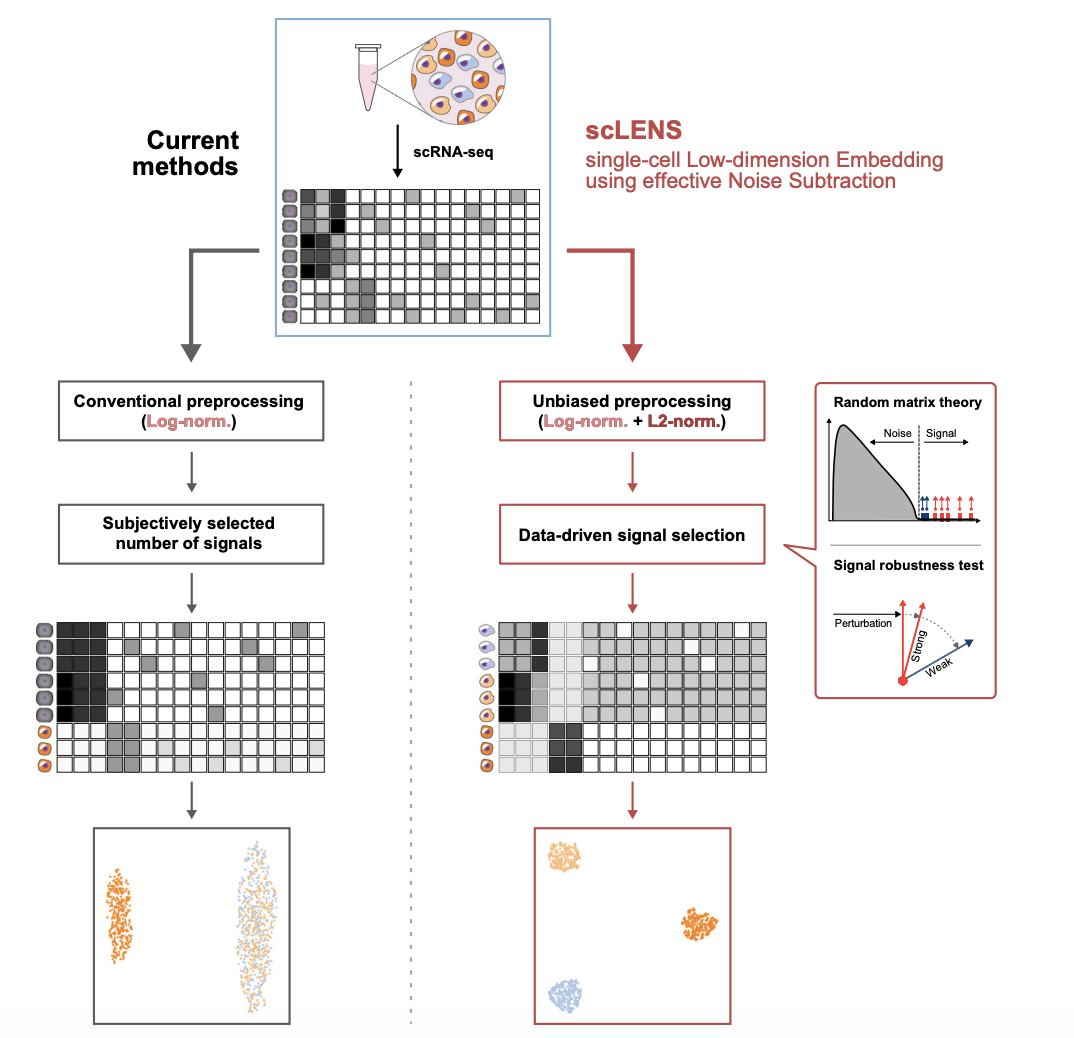 scLENS: data-driven signal detection for unbiased scRNA-seq data analysisNature Communications, 2024
scLENS: data-driven signal detection for unbiased scRNA-seq data analysisNature Communications, 2024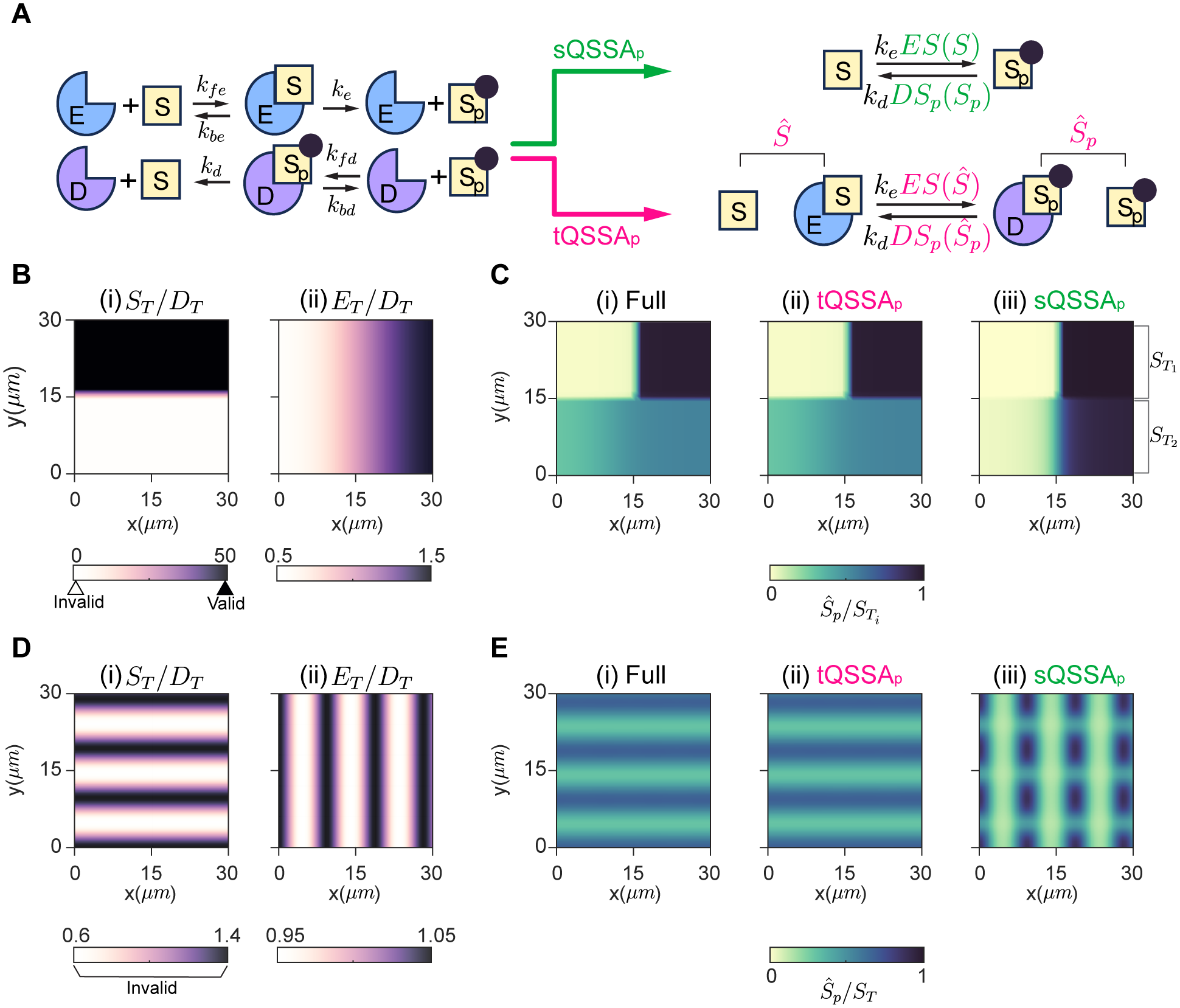 Beyond homogeneity: Assessing the validity of the Michaelis–Menten rate law in spatially heterogeneous environmentsPLOS Computational Biology, 2024
Beyond homogeneity: Assessing the validity of the Michaelis–Menten rate law in spatially heterogeneous environmentsPLOS Computational Biology, 2024




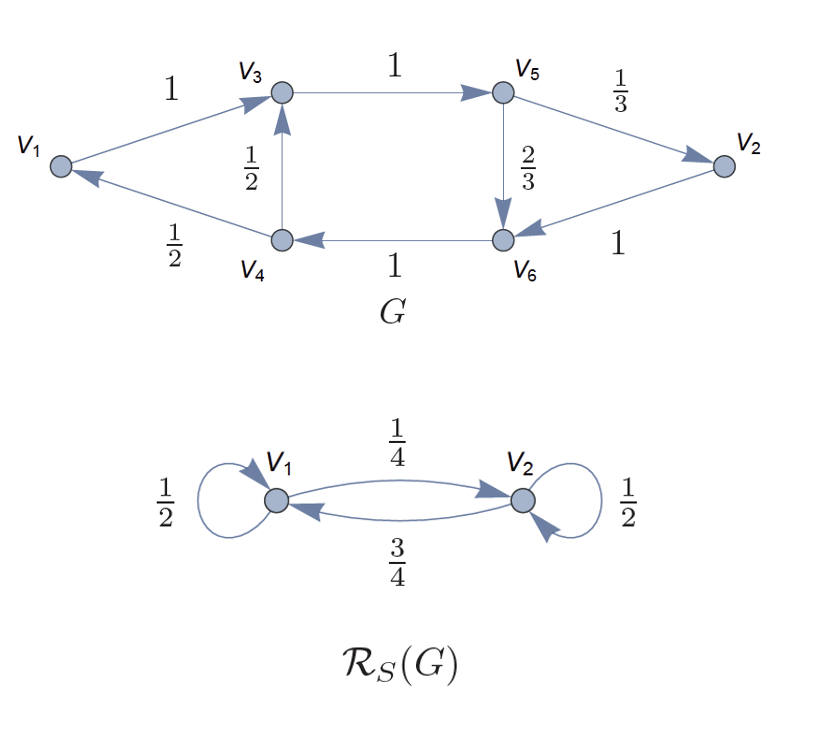 Uncovering Hierarchical Structure in Social Networks Using Isosxpectral ReductionsIn 2018 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining (ASONAM), 2018
Uncovering Hierarchical Structure in Social Networks Using Isosxpectral ReductionsIn 2018 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining (ASONAM), 2018